Phyloformer: towards fast and accurate phylogeny estimation with self-attention networks
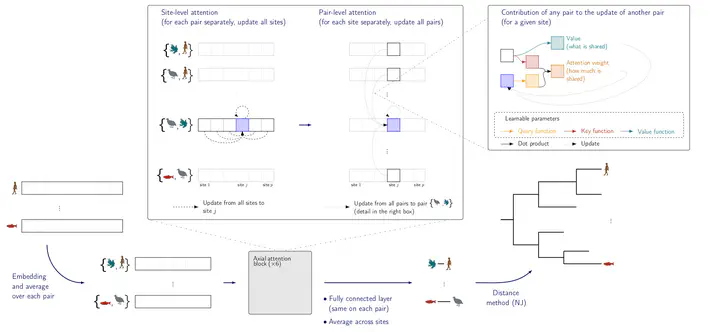 Image credit: Laurent Jacob
Image credit: Laurent JacobAbstract
An important problem in molecular evolution is that of phylogenetic reconstruction, that is, given a set of sequences descending from a common ancestor, the reconstruction of the binary tree describing their evolution from the latter. State-of-the-art methods for the task, namely Maximum likelihood and Bayesian inference, have a high computational cost, which limits their usability on large datasets. Recently researchers have begun investigating deep learning approaches to the problem but so far these attempts have been limited to the reconstruction of quartet tree topologies, addressing phylogenetic reconstruction as a classification problem. We present here a radically different approach with a transformer-based network architecture that, given a multiple sequence alignment, predicts all the pairwise evolutionary distances between the sequences, which in turn allow us to accurately reconstruct the tree topology with standard distance-based algorithms. The architecture and its high degree of parameter sharing allow us to apply the same network to alignments of arbitrary size, both in the number of sequences and in their length. We evaluate our network Phyloformer on two types of simulations and find that its accuracy matches that of a Maximum Likelihood method on datasets that resemble training data, while being significantly faster.