 Image credit: Dexiong Chen
Image credit: Dexiong ChenAbstract
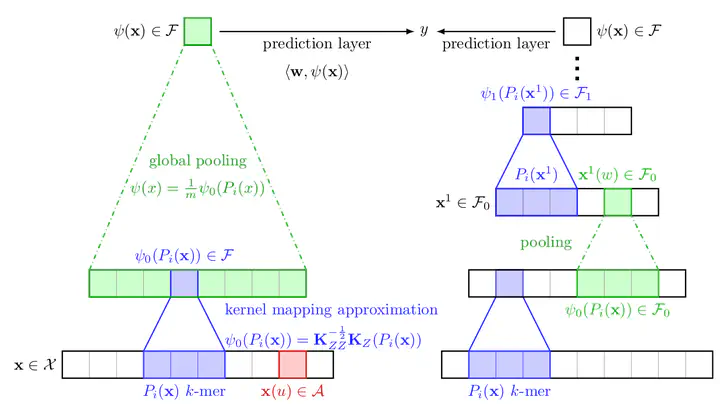
The growing number of annotated biological sequences available makes it possible to learn genotype-phenotype relationships from data with increasingly high accuracy. When large quantities of labeled samples are available for training a model, convolutional neural networks can be used to predict the phenotype of unannotated sequences with good accuracy. Unfortunately, their performance with medium- or small-scale datasets is mitigated, which requires inventing new data-efficient approaches. We introduce a hybrid approach between convolutional neural networks and kernel methods to model biological sequences. Our method enjoys the ability of convolutional neural networks to learn data representations that are adapted to a specific task, while the kernel point of view yields algorithms that perform significantly better when the amount of training data is small. We illustrate these advantages for transcription factor binding prediction and protein homology detection, and we demonstrate that our model is also simple to interpret, which is crucial for discovering predictive motifs in sequences.